The Polymerase Chain Reaction
Principle of the method
The polymerase chain reaction, PCR, is an extremely powerful technique
used for many purposes in molecular biology. It allows you to
produce copies of any specific segment from a complex mixture
of DNA. In a chain reaction copies are themselves copied, so that
the total number of product molecules increases exponentially
with time. Thus, a segment from a single molecule of DNA can be
amplified in an hour or two to produce many millions of copies,
which can then be detected and studied with standard methods.
Soon after its invention in 1983, it became a central method in the field of molecular
biology. It is now a mini-industry, with many companies selling
reagents and thermocyclers, at least 63 books in print (as of
1998) that describe its history and uses, and a journal entirely
devoted to results and variations of the method.
|
|
|
|
|
|
|
|
The ends of the DNA segment to be copied are determined by the
nucleotide sequence of the two primer oligonucleotides which are
added to the reaction. The double stranded sample DNA is first
denatured to produce two single strands, and the two primers then
hybridize to specific sequences on the two DNA complimentary strands.
The enzyme, DNA polymerase, then binds to the 3' ends of the hybridized
primers and extends them by adding nucleotides that are complementary
to the original DNA. In the first round of synthesis the new chains
grow as long as the enzyme can proceed in the time allotted for
the reaction. However, when these chains are copied in the next
round, the polymerase comes to the end of the chain and stops,
producing molecules of a fixed length. In the succeeding rounds
of amplification, the fixed length molecules become the predominant
species.
| |
|
|
 |
|
Uses of the PCR
|
|
|
|
|
The widespread use of the PCR depends on the ability to easily
synthesize or cheaply purchase oligonucleotides of any arbitrary
sequence 15-30 nucleotides long. Several companies sell automated
oligonucleotide synthesizers and associated reagents, and an oligonucleotide
can be produce in about an hour. You can purchase custom made
primers at about $1/base, with a 48 hour turnaround time. |
|
|
|
 |
|
To test for a mutant sequence: If the nucleotide sequence of the wild type and mutant gene are
known, you use one primer which will hybridize at the site of
the mutation if the wild type sequence is present. The second
primer should hybridize a convenient distance away, say 200 nucleotides.
If the sequence is wild type, a 200 nucleotide fragment will be
made. If the mutant sequence is present, the first primer will
not hybridize completely, and the 200 nucleotide fragment will
not be made. The first primer is usually constructed so the mutant
site is at the 3' end, since correct hybridization at this position
has the greatest effect on the ability of the polymerase to extend
the chain. |
|
|
|
|
|
|
|
|
To substitute for a bacterial or viral clone: If a genomic sequence was to be studied, saved, or transmitted
to another lab, it was typically cloned into a vector, which could
then be grown into a large culture. The sequence to be cloned
was defined by the presence of unique nuclease restriction sites
at each end. Finding unique restriction sites could be difficult.
With PCR, you need only to specify sequences about 25 nucleotides
long at each end. Anyone can then make or buy primers and make
as many copies as they need directly from genomic DNA. |
|
|
To determine the activity of a gene: Just measure the amount of mRNA present in the cell. Copy the
RNA into DNA, either with a separate polymerase, or use a DNA
polymerase that will use RNA as a template in the presence of
Mn++, e.g. Tth polymerase. Then amplify the DNA copy so you can
easily measure it. |
|
|
|
|
There are many other uses of PCR, (I haven't even mentioned forensics) and it must be used directly
or indirectly in half of the published studies that use techniques
of molecular biology. |
|
The DNA polymerases
Many different DNA polymerases, usually supplied by commercial firms, are used today in the
PCR. The enzymes are typically purified from E. coli containing
a polymerase gene from a thermophilic organism, introduce by recombinant
techniques. Some enzymes are particularly good at copying long
sequences, some have especially high fidelity, some copy RNA as
well as DNA, some cost less, some companies give you a T-shirt
if you buy enough, ...
Relation between the amount of product made and amount of starting
template
In many cases the goal of PCR is to produce material for another
purpose, or you just want to know if the target sequence is present
or not present in the original DNA sample. However, in some cases
you may want to determine the quantitative concentration of the
target sequence from the amount of product made by PCR. If the
efficiency of each and every round of the reaction was exactly
2.000000, there would be a one-to-one relation of product to starting
template. However, there isn't, and the lack of a direct proportionality
can be artificially divided into two categories.
|
|
|
|
|
|
|
A. Efficiency is a constant, but less than 1: If the fraction of target sequences copied at each cycle is the
efficiency, E, then the amount of product produced at the end
of N cycles, Y, is:
Y = (1 + E)N
When E is 1, you get a little over 109 copies after 30 cycles (the green line on the graphs). If E is
less than one, you still get exponential amplification, and thus
a straight line on a semi-log plot. However, if the efficiency
drops a mere 1 percent, so that E becomes 0.99, you get only 86
percent as much product after 30 cycles, a decrease of 14 percent!
B. Efficiency is 1 in early cycles, but then decreases: This produces a curved line in a semi-log plot, which may look
linear over a several cycles in a linear plot. If the efficiency
becomes very low at the end, the amount of product becomes almost
independent of the amount of starting material; the amplification
process has become saturated. In this situation it is difficult
to get much information about the number of copies of the amplified
sequence at the start of the PCR. |
|
 |
How can you determine the original concentration of amplified
sequences?
A. Measure product at a low concentration, so there is a strong
and predictable relation between product and sample levels:
|
|
|
|
|
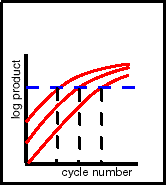
In the Figure to the left, the desired final level is the horizontal,
blue dashed line. However, since you don't know the concentration
of the sample when you start the PCR (after all, that's the purpose
of the assay), you don't know how many cycles of amplification
will be required (the 3 vertical, black dashed lines in the Figure
corresponding to the 3 input amounts).
1st Solution: Start with several identical reaction mixes, then
terminate the PCR and measure product level after different numbers
of amplification cycles. This increases the amount of work per
assay considerably.
2nd solution: Use a device that measures the amount of product in the tube after each PCR
cycle, i.e. without opening the tube. This requires that you purchase
such a device. |
|
|
|
|
 |
B. Add a known amount of a DNA standard to the sample, and measure
the ratio of sample to standard after the PCR has amplified both:
|
|
|
|
|
Since you want the standard to be amplified with an efficiency
as close as possible to the sample, the standard must be similar
to the sample, but still distinguishable from it. The DNA standard
should have the same size and end sequences as the sample sequence,
so that only one set of primers is needed But obviously it must
be different somewhere so its concentration can be measured independently
of the sample. The distinguishing sequence could be a single base
change, creating or eliminating a restriction site, or it could
be a bigger change that is detected by a hybridizing probe. |
|
|
|
|
 |
C. Construct a standard curve that relates amount of input template
to product:
|
|
|
|
|
The idea here is that you don't care what the quantitative relationship
is between input and output, as long as you know what it is, i.e.
have a standard curve. There are at least two weaknesses of this
approach.
1. The parameters that can affect efficiency are so numerous that
it is difficult to have confidence in controlling or even keeping
them constant from assay to assay.
2. If the amplification reaction is saturated, so that large changes
in input result in only small changes in output, accuracy is going
to be sacrificed, no matter how well you can predict the reaction.
|
|
A quantitative model of the PCR may be of some interest, if only
because it makes us think about mechanism and thus how PCR might
be improved. Typically, accumulation of product changes from an
exponential to linear at higher cycle numbers. It seems likely
that this occurs when the amount of product exceeds polymerase. Recent publications by Schnell
and Mendoza describe one model. A graph of amount of product after each amplification cycle,
for various values of Km (the only adjustable parameter of their
model), can be obtained by this Java implemented calculator. |
Return